








Who is the TGA?
In Australia, the Therapeutic Goods Administration (TGA) is the national regulator for medical devices and other therapeutic goods. It operates under the Department of Health and Aged Care and plays a key role in safeguarding public health.
The TGA is responsible for evaluating, licensing, and monitoring all medical devices to ensure they are safe, effective, and high quality before reaching the Australian market.
Why does the TGA matter for licensing medical devices?
If you’re planning to supply medical devices in Australia, TGA approval is essential. Medical devices must generally be included on the Australian Register of Therapeutic Goods (ARTG) before they can be legally supplied in Australia, unless a specific exemption, exclusion, or special access pathway applies.
The TGA’s regulatory framework covers the full product lifecycle, from pre-market approvals to post-market surveillance. Their standards are internationally recognised, making TGA compliance not only a legal requirement but a mark of product credibility.
P.S.: Getting TGA approval is one part and having an ABN is another. Learn how to register your ABN and start operating legally in Australia.
How the TGA Classifies Medical Devices by Risk
When it comes to licensing medical devices in Australia, the TGA follows a strict risk-based classification system. The higher the potential risk to patients or users, the more rigorous the approval process.
Getting your classification right from the start is critical; it shapes your entire regulatory path, from documentation to fees. Misclassify it, and you could face delays or rejection.
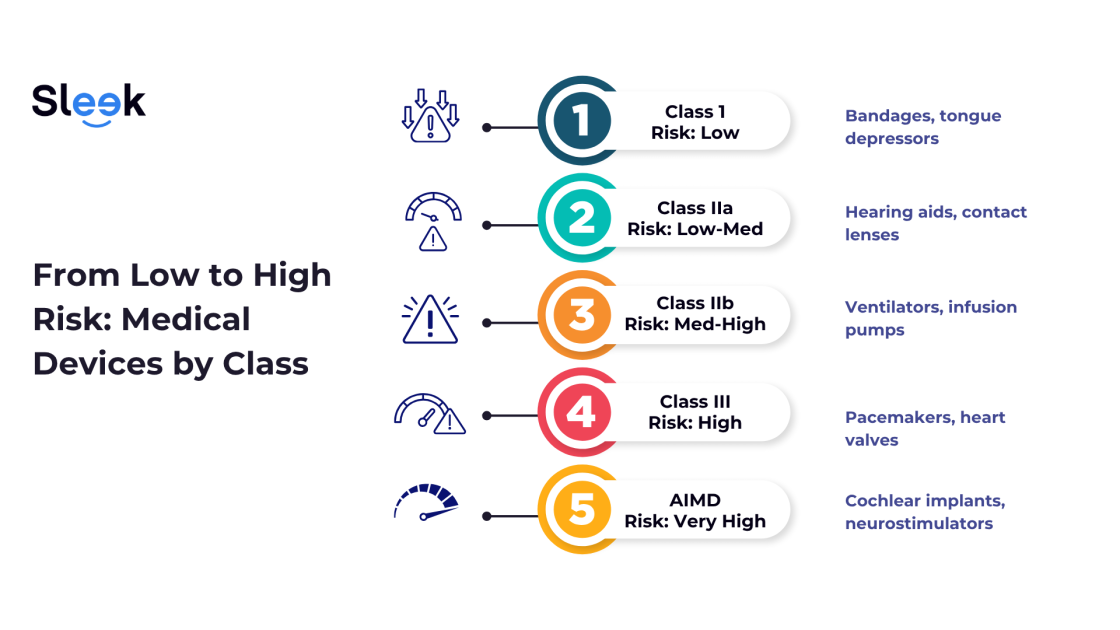
Here’s how the TGA breaks it down:
Class I: Low Risk
Basic, non-invasive items like bandages or tongue depressors.
Some may be Class Is (sterile) or Class Im (measuring), which have extra requirements.
Class IIa: Low to Medium Risk
Short-term or minimally invasive devices such as hearing aids or contact lenses.
Requires more technical documentation than Class I.
Class IIb: Medium to High Risk
Devices that support critical functions; think ventilators or infusion pumps.
Expect more in-depth assessments and evidence requirements.
Class III: High Risk
Implantable or life-sustaining devices like pacemakers or heart valves.
This class undergoes the most intensive scrutiny by the TGA.
AIMD: Active Implantable Medical Devices
Implantables that require a power source, like cochlear implants.
These sit at the highest end of the risk spectrum and face the strictest reviews.
Understanding where your device sits in this classification system is your first step toward getting it licensed and market-ready in Australia.
P.S.: The higher the risk class, the higher the stakes. Explore what business insurance you might need to stay protected.
TGA sponsor for licensing medical devices in Australia
If you’re based overseas and planning to license medical devices in Australia, you’ll need more than just a product, you’ll need a local representative. That’s where a TGA sponsor comes in.
What is a TGA sponsor?
A TGA sponsor is a registered Australian business or resident who takes legal responsibility for your device in Australia.
- They’re the ones who submit your application to the TGA, manage post-market reporting, and ensure your device stays compliant under Australian law.
- Their details appear on your ARTG certificate, so it’s a formal, accountable role, not just admin support.
From 2026, the TGA has reinforced sponsor responsibilities around importation definitions, documentation access, and ongoing compliance.
- Sponsors must ensure conformity assessment evidence and technical documentation are accessible within Australia.
- And that the device continues to meet the Essential Principles throughout its lifecycle.
Do you need a TGA sponsor to license medical devices?
Yes. If your company doesn’t have a legal presence in Australia, a TGA sponsor is mandatory. Without one, you can’t apply for TGA registration or supply your device locally.
Who can be a TGA sponsor?
The sponsor must be based in Australia, either an individual or an incorporated business. Some overseas companies use their distributor as a sponsor, but that approach can backfire.
Why an independent sponsor is often a smarter move
Your sponsor controls the ARTG listing. If things go sideways with your distributor, you don’t automatically keep your listing, they do. That means starting over, which wastes time, money, and market momentum.
Choosing an independent TGA sponsor gives you flexibility. You keep control of your regulatory pathway and can work with multiple distributors without risking your listing. It’s a cleaner, more scalable way to enter and grow in the Australian market.
P.S.: Looking to act as a sponsor or set up locally? Here’s what you need to know about getting an ACN in Australia.
Post-market obligations under TGA regulations (2026 Update)
TGA approval is not the end of your compliance obligations. Once your medical device is included in the ARTG, sponsors must meet ongoing post-market responsibilities.
These include:
Monitoring device performance and safety in the Australian market
Reporting adverse events and safety issues to the TGA within required timeframes
Maintaining access to up-to-date technical documentation and conformity evidence
Ensuring continued compliance with the Essential Principles
Complying with traceability and identification requirements where applicable (including Unique Device Identification obligations as progressively implemented)
In recent regulatory updates, the TGA has increased scrutiny on post-market vigilance and sponsor accountability. Failure to meet ongoing obligations can result in suspension, cancellation of ARTG inclusion, civil penalties, or enforceable undertakings.
Understanding these responsibilities early helps prevent costly compliance breaches and protects your long-term access to the Australian market.

What you’ll need to submit to the TGA?
To get your medical device listed on the ARTG, your submission needs to be complete, accurate, and audit-ready. While your TGA sponsor handles the actual submission, the manufacturer is responsible for supplying the documents.

Here’s what you’ll typically need:
- Manufacturer Evidence: Evidence of conformity assessment, such as an EU MDR certificate issued by a recognised Notified Body or certification from a Comparable Overseas Regulator (COR), where applicable. Manufacturer evidence must be submitted to and accepted by the TGA before ARTG inclusion.
- Declaration of Conformity: A legal statement confirming your device meets Australia’s Essential Principles and conformity assessment requirements.
- Labelling & IFU (Instructions for Use): Must comply with TGA standards, including sponsor details and safe-use instructions.
- Technical Documentation: A complete technical file demonstrating compliance with Australia’s Essential Principles. This includes risk management documentation (e.g. ISO 14971), clinical evidence (where required), design and manufacturing information, performance data, quality management system certification (e.g. ISO 13485), and post-market surveillance procedures. Documentation must be audit-ready.
- Device classification: The device must be correctly classified under Australian regulations (Class I, Is, Im, Ir, IIa, IIb, III, AIMD, or IVD classes). Documentation requirements and audit pathways depend on classification.
- GMDN Code: A valid and accurate Global Medical Device Nomenclature (GMDN) code must be selected. The code must precisely match the device’s intended purpose. Incorrect selection may result in delays, audit findings, or rejection of the application.
Getting this right upfront helps avoid delays and smooths your path to market approval.
What is a GMDN code and why does it matter?
The GMDN (Global Medical Device Nomenclature) code is an internationally recognised system used to classify medical devices. Each device must be assigned the correct GMDN code as part of your TGA application.
This code:
- Helps the TGA categorise and track your device
- Links your product to safety and performance data globally
- Must accurately reflect the intended use and function of your device
Choosing the right GMDN code is key. If it doesn’t match your product, your application may be delayed or rejected. Your sponsor or regulatory consultant can help confirm the best match.



